
目录
声子是晶体中晶体结构集体激发的准粒子。它是晶格集体振动模式量子化的描述,满足玻色-爱因斯坦统计,与晶体的热力学性质有着密切的关系。因此计算声子的色散关系,也就是声子谱显的尤为重要。过程是通过第一性原理得到所需要的力常数数据,后使用phonopy进行辅助处理和计算。第一性常见的两种方法分别是:有限位移法 (finite displacement method ) 和 密度泛函微扰理论 (Density functional perturbation theory, DFPT) 。
所需工具
在计算之前所需要准备的一些软件
- VASP (需要
5.3及以上的版本,我使用的是组里购买的6.1.1版本) - phonopy (因为后处理中分析振动模式太高版本的不支持,所以我使用的是v2.4.2 版本)
- VESTA (不必须,但是便于模型可视化)
- vaspkit (VASP计算的后处理软件,十分实用,但在计算声子谱过程中也不必须)
另外介绍两个有用网站
- SAM (分析红外和拉曼模式)
- Phonon website (十分实用的可视化分析声子振动模式,尤其是现在v_sim官网无法访问)
概述
Phonopy软件提供了一个简单的从VASP输出结果中提取晶格振动和热力学性质,在这个教程里是展示通过VASP和phonopy得到声子的色散,态密度和提取声子的振动模式分析。在这里我主要以单层为例子,进行过程的具体阐述。一般而言,计算分为以下几个步骤:
声子谱计算步骤
- 优化晶格结构和原子位置,检查原子的位置和晶格常数,保证晶格原有的对称性;
- 通过有限位移法或者或者密度泛函微扰理论 (DFPT) 计算;
- 通过phononpy进行后处理分析。例如声子谱,态密度,振动模式等等。
结构优化
为了得到准确的声振动模式,我们首先需要的是对单层晶格结构进行优化。VASP通过ISIF标签[1]提供了不同的结构优化的方法。常用的分为两种不同的情况[2]:
- 已知晶格原有的对称性,但是晶格常数和内部原子的位置并不知道。在这样的情况下,需要保证
POSCAR文件中的初始结构具有正确的对称性,而为了保证这种对称性不变需要设置:ISYM=2和使用IBRION=2的优化算法。同时需要对ISIF进行确定。提示
ISIF=2: 保持晶格常数不变优化原子位置;ISIF=3: 晶格常数和原子位置都在发生改变,同时体积也在发生改变;ISIF=4: 晶格常数和原子位置都在发生改变,但是体积不发生改变;
- 初始结构对称性的对称性并不是正确的,这样的情况下,需要关闭对称性而进行继续:
ISYM=0和使用IBRION=2的优化算法。而同时一般需要ISIF=3。
但是在计算之后当你查看CONTCAR文件的输出文件时,你注意到晶格的对称性可能已经改变,而缺乏对称性或者对称性不正确会导致错误的认识声子的振动模式,因此为了修复这个问题,你需要对于CONTCAR文件进行编辑,在晶格矢量中看到一些很小的趋于零的值,如-0.00001248932473,你应该把它修改为 0.00000000000000,之后再设置ISIF=2保持晶格常数不变保持对称性设置ISYM=2。之后得到的结构就会含有你想要去计算声子的对称性。
对单层晶格结构(单层铁磁态),原胞结构如图所示
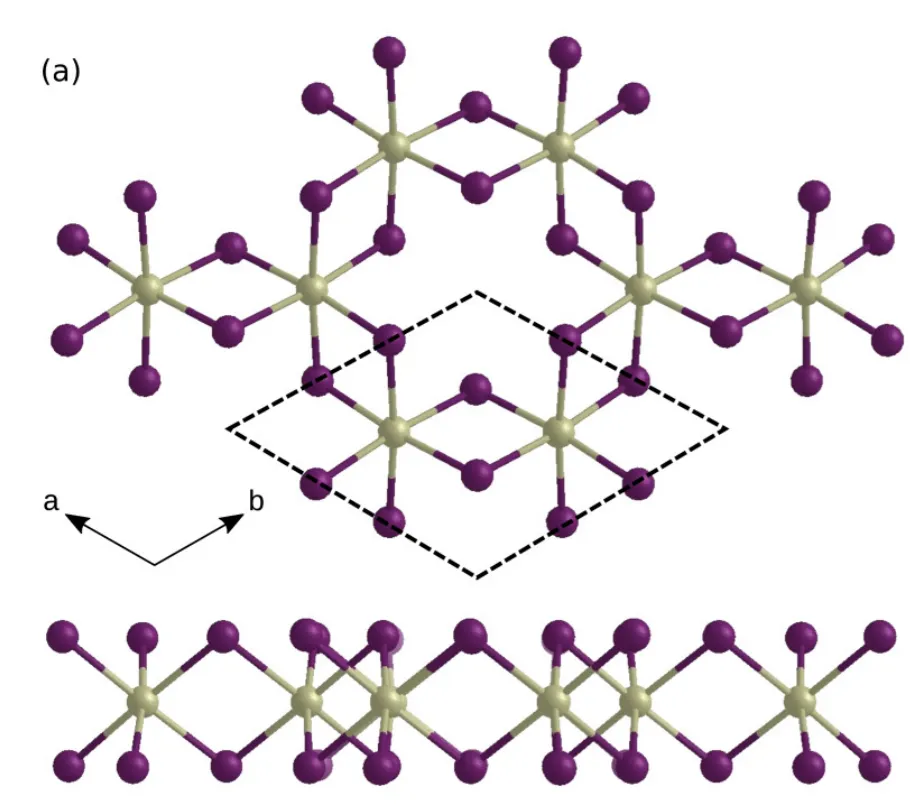
结构具有的对称性:点群是 ,空间群是 (162)。
初始结构的POSCAR文件
POSCARCrI3_sym 1.0 7.0019998550 0.0000000000 0.0000000000 -3.5009999275 6.0639097518 0.0000000000 0.0000000000 0.0000000000 23.1219997406 Cr I 2 6 Direct 0.333333343 0.666666687 0.500000000 0.666666627 0.333333313 0.500000000 0.666666687 0.000000000 0.567520022 0.333333313 0.000000000 0.432479978 0.000000000 0.666666687 0.567520022 0.000000000 0.333333313 0.432479978 0.333333313 0.333333313 0.567520022 0.666666687 0.666666687 0.432479978
在这里是已知了我们需要的对称性,所以采用第一种情况的方法进行优化,先进行ISIF=3和ISYM=2的计算,INCAR文件
bashSYSTEM = monolayer_CrI3
ENCUT = 600
ISTART = 0
ICHARG = 3
ISMEAR = 0; SIGMA=0.05
NPAR = 2
PREC = Accurate
#relaxation
ISIF = 2
NSW = 60
NELM = 100
NELMIN = 5
EDIFF = 1E-7
EDIFFG = -0.002 #这里设置的值是因为所有力的收敛值在0.001振荡
IBRION = 2
IALGO = 38
#magnetism
ISPIN=2
MAGMOM = 3.0 3.0 6*0
#symmetry
ISYM = 2
得到晶格常数的CONTCAR文件
POSCARCrI3_sym 1.00000000000000 7.0004768389826655 -0.0000000000000000 0.0000000000000000 -3.5002384194913327 6.0625907812382094 0.0000000000000000 -0.0000000000000000 0.0000000000000000 23.0926512132158557 Cr I 2 6 Direct 0.3333333429999996 0.6666666870000029 0.5000000000000000 0.6666666269999979 0.3333333129999971 0.5000000000000000 0.6400705371735319 -0.0000000000000000 0.5676041836998535 0.3599294628264680 -0.0000000000000000 0.4323958163001465 -0.0000000000000014 0.6400705371735294 0.5676041836998535 0.0000000000000014 0.3599294628264708 0.4323958163001465 0.3599294628264694 0.3599294628264708 0.5676041836998535 0.6400705371735306 0.6400705371735294 0.4323958163001465
可以使用vaspkit分析弛豫之后的结构的对称性
bash-->> (01) Reading Structural Parameters from CONTCAR File... +-------------------------- Summary ----------------------------+ Prototype: AB3 Total Atoms: 8 Formula Unit: CrI3 [ Alphabetically Listed: CrI3 ] Full Formula Unit: Cr2I6 Crystal System: Trigonal Crystal Class: -3m Bravais Lattice: hP Lattice Constants: 7.000 7.000 23.093 Lattice Angles: 90.000 90.000 120.000 Volume: 980.076 Density (g/cm3): 1.466 Space Group: 162 Point Group: 20 [ D3d ] International: P-31m Symmetry Operations: 12 +---------------------------------------------------------------+
依然保持原有的对称性,因此可以用来计算声子谱。
力常数计算
如之前所说的力常数的计算可以有两种方法:有限位移法 (finite displacement method ) 和 密度泛函微扰理论 (Density functional perturbation theory, DFPT) 。在我的计算中使用的是DFPT的方法,我也会在这里介绍有限位移法的计算过程。
前期预处理
把上述的POSCAR文件拷贝成POSCAR-unitcell文件。建立超胞结构,由于是单层结构,所以我这里建立的是的超胞结构
bashphonopy -d --dim="2 2 1" -c POSCAR-unitcell
可以看到目录下生成了三类文件
SPOSCAR(超胞的结构文件);POSCAR-001,POSCAR-002, ,POSCAR-{number}(在某些方向对某些原子进行了微小的位移后的POSCAR文件);phonopy_disp.yaml(记录建立超胞时不同原子位移的信息)。
密度泛函微扰理论
将SPOSCAR 文件重新命名为POSCAR,之后运行VASP计算,其中注意的是使用的参数是IBRION=8和NSW=1,计算单层具体的INCAR文件设置如下
bashSYSTEM = monolayer_CrI3
ENCUT = 600
IBRION = 8
NSW = 1
ISMEAR = 0; SIGMA = 0.01
PREC = A
IALGO = 38
NELM = 120
SYMPREC = 5E-4
EDIFF = 1E-7
LWAVE = F
LCHARG = F
LREAL = F
ADDGRID = T
LASPH = T
#Magnetism
ISPIN=2
MAGMOM = 3.0 3.0 3.0 3.0 3.0 3.0 3.0 3.0 24*0
之后通过VASP计算得到的vasprun.xml文件,从这个文件中得到力常数FORCE_CONSTANTS文件。
bashphonopy --fc vasprun.xml
有限位移法
分别将POSCAR-{number}文件作为VASP的计算的POSCAR文件,因此建立不同的子文件夹,比如命名为disp-{number},分别将POSCAR-{number} 拷贝进文件夹内,同时分别准备好KPOINTS和POTCAR文件,另外INCAR文件的设置为
SYSTEM = monolayer_CrI3 ENCUT = 600 IBRION = -1 ISMEAR = 0; SIGMA = 0.01 PREC = A IALGO = 38 NELM = 120 NPAR = 2 EDIFF = 1E-7 LWAVE = F LCHARG = F LREAL = F #Magnetism ISPIN=2 MAGMOM = 3.0 3.0 3.0 3.0 3.0 3.0 3.0 3.0 24*0
因为每一个POSCAR-{number}都需要建立一个子文件夹,在这里为了方便计算过程本人写了一个简单的shell脚本
bash#!/bin/bash
for x in 1 2 3 4 5
do
echo "when Index = " $x
if [ $x -lt 10 ]
then
index=00$x
else
Emin=0$x
fi
rm -rf disp-${index}/
mkdir disp-${index}/
cp POSCAR-${index} disp-${index}/POSCAR
cp job.pbs INCAR POTCAR KPOINTS disp-${index}/
cd disp-${index}/
qsub job.pbs > number
echo "qsub job.pbs > number"
cd ../
done
解释一下上面的脚本
- 第二行中{1 2 3 4 5} 是有多少个
POSCAR-{number}就从写到number; job.pbs是提交服务器计算的pbs脚本,根据自己的情况自行更改;- 程序也有不足的的地方,如number不能大于100(更换其中的if语句,但是一般不会有这么多)。
计算之后可以通过每个子文件夹中的vasprun.xml文件提取出FORCE_SETS
bashphonopy -f disp-001/vasprun.xml disp-002/vasprun.xml disp-003/vasprun.xml ... disp-{number}/vasprun.xml
或者
bashphonopy -f disp-{001...{number}}/vasprun.xml
后处理
通过phononpy进行后处理分析。例如声子谱,态密度,振动模式等等。在这里我是读取用DFPT得到的FORCE_CONSTANTS文件,所以再phonopy的控制输入文件中需要加入FORCE_CONSTANTS = READ或者运行phonopy是加上--readfc选项,而对于读取有限位移法得到的FORCE_SETS文件却不需要。
声子谱
声子谱的phonoy的输入文件{name}.conf,{name}可以随意命名,如band.conf如下
bashFORCE_CONSTANTS = READ ATOM_NAME = Cr I DIM = 2 2 1 NPOINTS = 101 BAND = 0.000000 0.000000 0.000000 0.500000 0.000000 0.000000 0.333333 0.333333 0.000000 0.000000 0.000000 0.000000 BAND_LABELS = $\Gamma$ M K $\Gamma$ EIGENVECTORS = .TRUE
运行
phonopy -p -s band.conf -c POSCAR-unitcell
其中-p是指定输入文件band.conf,-s是指定输出的文件是pdf格式,而-c是指定输入的原胞文件是POSCAR-unitcell(如果原胞的文件是POSCAR,则不需要指定)。则得到铁磁态的单层的声子谱
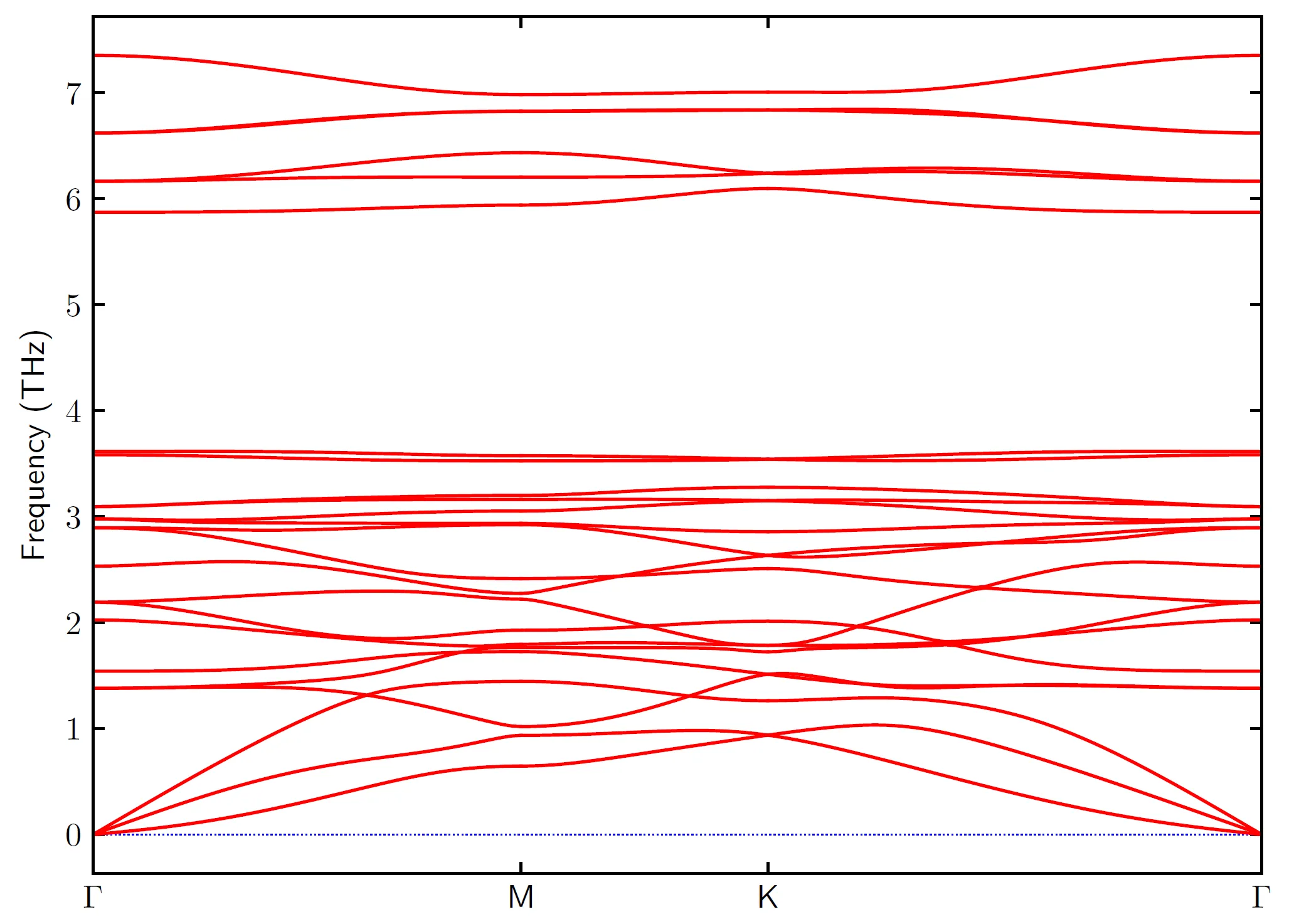
为了导出声子谱的数据,其中数据是写在band.yaml文件中,我自己写了一个简单的python文件,将声子谱的数据写入band.dat文件,程序如下:
pythonimport yaml
import numpy as np
with open("band.yaml", 'r') as stream:
data = yaml.safe_load(stream)
phon = data['phonon']
Natom = data['natom']
print('Number of atoms:',Natom)
Nkp=len(phon)
print('Number of k-points:',Nkp)
lines = []
for i in range(Nkp):
distance=phon[i]['distance']
band = phon[i]['band']
Nband=len(band)
lines.append("%8.5f\t" % distance)
for j in range(Nband):
eig=band[j]['frequency']
lines.append("%8.5f\t" % eig )
lines.append("\n" )
print('Number of bands:',Nband)
print("Begin write band.dat ...")
with open("band.dat",'w') as f:
f.writelines(lines)
print("Done!!")
态密度
态密度的phonopy的输入文件dos.conf,其中PDOS是投影到原子上的态密度
bashFORCE_CONSTANTS = READ
ATOM_NAME = Cr I
DIM = 2 2 1
MP = 64 64 1
DOS_RANGE = -1 8 0.01 #能量取值间隔和范围
PDOS = 1 2, 3 4 5 6 7 8 #投影态密度,Cr原子的态密度(1,2)和I原子态密度分布(3-8)
同理运行
bashphonopy -p -s dos.conf -c POSCAR-unitcell
和得到声子一样,将会生成total_dos.pdf和partial_dos.pdf 图像文件。同时会导出数据文件total_dos.dat和projected_dos.dat ,下面是我通过这些数据绘制出来的图。
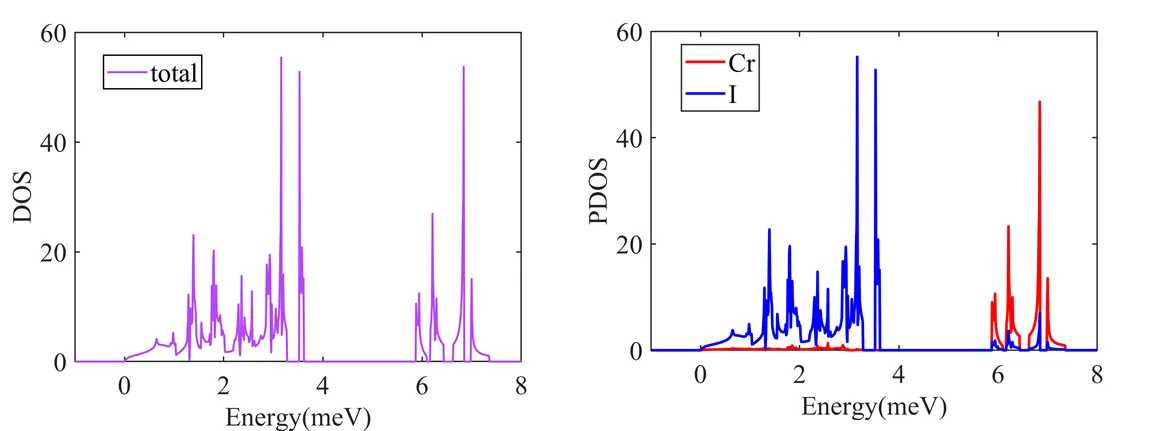
可以看出原子的振动模式主要分布在低频率,而原子的振动模式分布在高频率,这是因为原子比原子要轻许多。
振动模式
为了分析声子谱的振动模式,也就是各个点下每个频率下的本征矢量。在这里我推荐一个十分实用的分析振动模式的网站,就是在开头所需工具中提及的Phonon website 和SAM 。自己通过浏览相应的网站就知道如何使用了。
同时为了方便自己分析,和之前声子谱一样将本征矢量输出到vec.dat文件中,程序参考如下:
pythonimport yaml
import math
import numpy as np
with open("band.yaml", 'r') as stream:
data = yaml.safe_load(stream)
phon = data['phonon']
Natom = data['natom']
print('Number of atoms:',Natom)
Nkp=len(phon)
print('Number of k-points:',Nkp)
lines = []
for i in range(Nkp):
distance=phon[i]['distance']
band = phon[i]['band']
Nband=len(band)
for j in range(Nband):
eig=band[j]['frequency']
vec=band[j]['eigenvector']
for k in range(Natom):
atomvec=vec[k]
lines.append("%8.5f\t" % distance)
lines.append("%8.5f\t" % eig )
lines.append("%-5d\t" % (k+1))
lines.append("%15.10f\t" % atomvec[0][0])
lines.append("%15.10f\t" % atomvec[0][1])
lines.append("%15.10f\t" % atomvec[1][0])
lines.append("%15.10f\t" % atomvec[1][1])
lines.append("%15.10f\t" % atomvec[2][0])
lines.append("%15.10f\n" % atomvec[2][1])
print('Number of bands:',Nband)
print("Begin write band.dat ...")
with open("vec.dat",'w') as f:
f.writelines(lines)
print("Done!!")
参考
[1] vasp的ISIF标签
本文作者:sbyu
本文链接:
版权声明:本博客所有文章除特别声明外,均采用 BY-NC-SA 许可协议。转载请注明出处!